
Reviewed by Leslie Magtanong and Scott J. Dixon
A New Form of Cell Death
Cell death is a critical process required for normal development (e.g., removal of interdigital webs) and the maintenance of proper homeostasis throughout an organism’s lifetime (e.g., immune cell turnover). Cell death can be executed via caspase-mediated apoptosis or through one of a growing number of non-apoptotic cell death processes, including necroptosis, pyroptosis, parthanatos, and others.1 Ferroptosis is one of the more recently described forms of non-apoptotic cell death.2 This is an oxidative, iron-dependent process with cell death resulting from the accumulation of lipid reactive oxygen species (ROS) to toxic levels.3,4 Unlike apoptosis and other forms of non-apoptotic cell death, there is no evidence to date that the execution of ferroptosis requires one specific effector protein (e.g., a pore-forming protein). Rather, this lethal process is centered on the iron-dependent, oxidative destruction of membrane lipids.
As implied in its name, ferroptotic cell death is defined by the requirement for iron. Extracellular iron is bound by the protein transferrin, and internalized by the transferrin receptor.5,6 Iron is released from internalized transferrin/ transferrin receptor complexes from the lysosome into the cytoplasm.5,6 Within the cytosol, free iron can participate in Fenton chemistry to generate hydroxyl radicals. These radicals can then react with polyunsaturated acyl chains found in membrane phospholipids to generate lipid radicals that are easily attacked by oxygen to create highly reactive lipid peroxyl radicals. In turn, these radicals can abstract protons from neighboring phospholipids, leading to the generation of additional lipid peroxides and the propagation of damage throughout the membrane. This process of autoxidation may be sufficient to cause frank membrane permeabilization during ferroptosis in some cells,7 while in others it is likely that the enzymatic oxidation of PUFAs by iron-containing 12/15-lipoxygenase (LO; i.e., LOX) enzymes also contributes to this process.8-10
The Role of GPX4
Normally, highly reactive lipid peroxides are reduced to non-reactive lipid alcohols by the activity of the essential enzyme glutathione hydroperoxidase 4 (GPX4). GPX4 is a selenocysteine-containing enzyme that requires the reduced form of the antioxidant tripeptide glutathione (GSH) for its activity.11 Cysteine is the rate-limiting substrate in GSH biosynthesis, an ATP-dependent process wherein cysteine is conjugated to glutamate and glycine. Cysteine can be synthesized from methionine (via transsulfuration) but can also be imported into the cell as cystine (Cys2, the oxidized form of cysteine) through a sodium-independent antiporter called system xc-. System xc- is a heterodimer composed of xCT (encoded by SLC7A11), a heavy chain, and 4F2 (encoded by SLC3A2), a light chain.12 This antiporter exchanges intracellular glutamate for extracellular Cys2 in a one-to-one ratio. Once in the cytosol, cystine is reduced to cysteine, which enters the glutathione biosynthesis pathway. By maintaining the intracellular pool of cysteine, system xc- activity is a key node preventing the onset of ferroptosis in many cells. When GSH levels fall below a certain threshold, GPX4 is no longer active, lipid ROS accumulate to toxic levels and cells undergo ferroptotic cell death.2,13
 Ferroptosis can be induced by inhibiting the system xc--glutathione-GPX4 axis at various points. Small molecules that deplete glutathione levels are referred to as Class I ferroptosis-inducing compounds (FINs), while those that cause ferroptosis through direct inhibition of GPX4 without depleting glutathione are termed Class II FINs.14 The Class I FIN, erastin, was initially described to be selectively lethal to engineered tumor cells expressing oncogenic HRASV12.15,16 Erastin (and analogs), the tyrosine kinase inhibitor sorafenib (i.e., BAY 43-9006), and the antirheumatic drug sulfasalazine can all inhibit system xc-, deplete GSH, and trigger ferroptosis.2,17 Treatment with Class II FINs such as (1S,3R)-RSL3, ML-162, and related molecules covalently bind to and inactivate GPX4.8,14 The endoperoxide FINO2 promotes ferroptosis both by decreasing GPX4 activity and oxidizing ferrous iron.18 Finally, the oxime-containing molecule FIN56 induces ferroptosis by reducing GPX4 protein levels and also interfering with the synthesis of the endogenous lipophilic antioxidant metabolite coenzyme Q10 (CoQ10).19 Other molecules, like statins, may also induce ferroptosis indirectly by destabilizing GPX4 protein and inhibiting CoQ10 synthesis.20 While Class I FINs have shown some promise as inducers of ferroptosis in cancer cells in vivo, the development of analogs with greater potency in vivo remains a high priority.14,17,21 Inhibiting GPX4 has recently been pinpointed as a means to selectively kill drug-resistance cancer cell sub-populations.20,22,23 However, to date, none of the existing Class II FINs have shown in vivo activity. The development of such agents will be necessary to further investigate the potential for targeting GPX4 in cancer in vivo. Other agents, such as iron-binding nanoparticles, may provide an alternative means to induce ferroptosis in vivo.24
Ferroptosis can be induced by inhibiting the system xc--glutathione-GPX4 axis at various points. Small molecules that deplete glutathione levels are referred to as Class I ferroptosis-inducing compounds (FINs), while those that cause ferroptosis through direct inhibition of GPX4 without depleting glutathione are termed Class II FINs.14 The Class I FIN, erastin, was initially described to be selectively lethal to engineered tumor cells expressing oncogenic HRASV12.15,16 Erastin (and analogs), the tyrosine kinase inhibitor sorafenib (i.e., BAY 43-9006), and the antirheumatic drug sulfasalazine can all inhibit system xc-, deplete GSH, and trigger ferroptosis.2,17 Treatment with Class II FINs such as (1S,3R)-RSL3, ML-162, and related molecules covalently bind to and inactivate GPX4.8,14 The endoperoxide FINO2 promotes ferroptosis both by decreasing GPX4 activity and oxidizing ferrous iron.18 Finally, the oxime-containing molecule FIN56 induces ferroptosis by reducing GPX4 protein levels and also interfering with the synthesis of the endogenous lipophilic antioxidant metabolite coenzyme Q10 (CoQ10).19 Other molecules, like statins, may also induce ferroptosis indirectly by destabilizing GPX4 protein and inhibiting CoQ10 synthesis.20 While Class I FINs have shown some promise as inducers of ferroptosis in cancer cells in vivo, the development of analogs with greater potency in vivo remains a high priority.14,17,21 Inhibiting GPX4 has recently been pinpointed as a means to selectively kill drug-resistance cancer cell sub-populations.20,22,23 However, to date, none of the existing Class II FINs have shown in vivo activity. The development of such agents will be necessary to further investigate the potential for targeting GPX4 in cancer in vivo. Other agents, such as iron-binding nanoparticles, may provide an alternative means to induce ferroptosis in vivo.24
Ferroptosis Inhibition
Limiting the accumulation of lipid ROS prevents ferroptosis, even when GSH is depleted or GPX4 is inactivated. Given the central importance of iron and membrane polyunsaturated fatty acyls, mutations that disrupt either iron uptake or the incorporation of lipids into cellular membranes can both act as potent suppressors of ferroptosis. Thus, genetic silencing of the transferrin receptor limits iron accumulation and the induction of ferroptosis.25,26 Incorporation of iron into iron-sulfur clusters also serves as an endogenous mechanism of limiting free iron accumulation within the cytosol, and is especially important to limit ferroptosis during hypoxia.27 The incorporation of polyunsaturated fatty acids (PUFAs) into membrane lipids first requires the activation of the free fatty acids into PUFA-CoAs by acyl-CoA synthetase long chain family member 4 (ACSL4) and incorporation of the PUFA-CoA into lipids by lysophosphatidylcholine acyltransferase 3 (LPCAT3).2,28-30 Genetic perturbation of these enzymes can, therefore, block cell death under ferroptosis-inducing conditions by eliminating the key substrate (i.e., PUFA-containing lipids) required for the execution of this process. Natural product and synthetic small molecule iron chelators and lipophilic antioxidants are also effective inhibitors of cell death induced by both classes of FINs.4 Deferoxamine is a bacterial siderophore that chelates ferric iron. It was one of the first molecules found to inhibit ferroptosis.2,25 Lipophilic antioxidants such as ferrostatin-1 and liproxstatin-1 can also all block ferroptosis in vitro and, with varying degrees of potency, in vivo. 2,13,31,32 Structurally, ferrostatin-1 and liproxstatin-1 are both diarylamines. Mechanistically, their antioxidant activity stems from their ability to trap acyl chain-carrying peroxyl radicals in lipid bilayers.33-35 Other small molecules such as vitamin E hydroquinone may prevent ferroptosis by inhibiting the function of LO enzymes.36 Finally, interfering with the metabolic conversion of glutamine to glutamate using compound 968 can also attenuate ferroptosis in some models, but the biochemical mechanism connecting glutamine to iron and/or lipid ROS remains poorly defined.26 Nonetheless, these ferroptosis inhibitors have been demonstrated to prevent cellular damage and death in a wide range of ex vivo and in vivo models of acute and chronic pathological cell death,2,13,26,32 and testing in more complex diseases models or human clinical trials should be anticipated in the future.
Identify Novel Inducers of Ferroptosis
FSP1 Fluorescent Inhibitor Screening Assay Kit - NEW
Cayman's FSP1 Fluorescent Inhibitor Screening Assay Kit provides a robust and easy-to-use platform for identifying novel inhibitors of human FSP1, a negative regulator of the ferroptosis pathway.
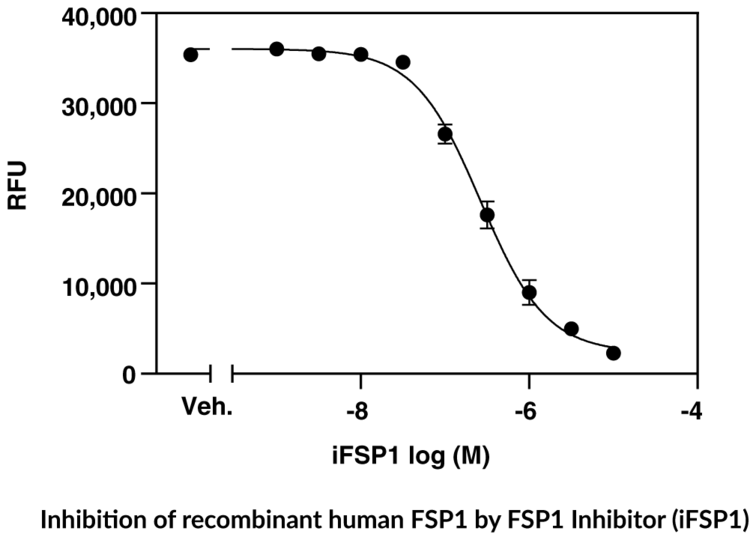
- Screen for inhibitors of FSP1
- Includes recombinant human FSP1 and positive control FSP1 inhibitor iFSP1
- Assay 45 samples in duplicate or 29 samples in triplicate
- Plate-based fluorometric measurement (Ex/Em = 540/590 nm)
GPX4 Inhibitor Screening Assay Kit - NEW
Cayman's GPX4 Inhibitor Screening Assay Kit provides a robust and easy-to-use platform for identifying novel inhibitors of human GPX4, a negative regulator of the ferroptosis pathway.
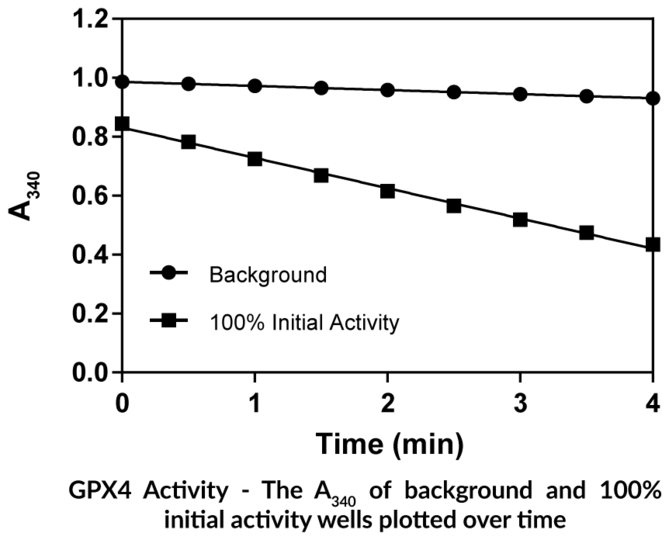
- Screen for inhibitors of GPX4
- Includes recombinant human GPX4
- Assay 45 samples in duplicate or 29 samples in triplicate
- Plate-based colorimetric measurement (340 nm)
References
- Galluzzi, L., Vitale, I., Aaronson, S.A., et al. Cell Death Differ. 25(3), 486-541 (2018).
- Dixon, S.J., Lemberg, K.M., Lamprecht, M.R., et al. Cell 149(5), 1060-1072 (2012).
- Cao, J.Y. and Dixon, S.J. Cell. Mol. Life Sci. 73(11-12), 2195-2209 (2016).
- Stockwell, B.R., Friedmann Angeli, J.P., Bayir, H., et al. Cell 171(2), 273-285 (2017).
- Andrews, N.C. and Schmidt, P.J. Annu. Rev. Physiol. 69(1), 69-85 (2007).
- Kawabata, H. Free Radic. Biol. Med. (2018).
- Shah, R., Shchepinov, M.S., and Pratt, D.A. ACS Cent. Sci. 4(3), 387-396 (2018).
- Yang, W.S., Kim, K.J., Gaschler, M.M., et al. Proc. Natl. Acad. Sci. U.S.A. 113(34), E4966-E4975 (2016).
- Shintoku, R., Takigawa, Y., Yamada, K., et al. Cancer Sci. 108(11), 2187-2194 (2017).
- Wenzel, S.E., Tyurina, Y.Y., Zhao, J., et al. Cell 171(3), 628-641 (2017).
- Ingold, I., Berndt, C., Schmitt, S., et al. Cell 172(3), 409-422 (2018).
- Lewerenz, J., Hewett, S.J., Huang, Y., et al. Antioxid. Redox Signal. 18(5), 522-555 (2013).
- Skouta, R., Dixon, S.J., Wang, J., et al. J. Am. Chem. Soc. 136(12), 4551-4556 (2014).
- Yang, W.S., SriRamaratnam, R., Welsch, M.E., et al. Cell 156(1-2), 317-331 (2014).
- Dolma, S., Lessnick, S.L., Hahn, W.C., et al. Cancer Cell 3(3), 285-296 (2003).
- Yagoda, N., von Rechenberg, M., Zaganjor, E., et al. Nature 447(7146), 864-868 (2007).
- Dixon, S.J., Patel, D.N., Welsch, M., et al. Elife 3, e02523 (2014).
- Gaschler, M.M., Andia, A.A., Liu, H., et al. Nat. Chem. Biol. 14(5), 507-515 (2018).
- Shimada, K., Skouta, R., Kaplan, A., et al. Nat. Chem. Biol. 12(7), 497–503 (2016).
- Viswanathan, V.S., Ryan, M.J., Dhruv, H.D., et al. Nature 547(7664), 453-457 (2017).
- Larraufie, M.-H., Yang, W.S., Jiang, E., et al. Bioorg. Med. Chem. Lett. 25(21), 4787-4792 (2015).
- Tsoi, J., Robert, L., Paraiso, K., et al. Cancer Cell 33(5), 890-904 (2018).
- Hangauer, M.J., Viswanathan, V.S., Ryan, M.J., et al. Nature 551(7679), 247-250 (2017).
- Kim, S.E., Zhang, L., Ma, K., et al. Nat. Nanotechnol. 11(11), 977-985 (2016).
- Yang, W.S. and Stockwell, B.R. Chem. Biol. 15(3), 234-245 (2008).
- Gao, M., Monian, P., Quadri, N., et al. Mol. Cell 59(2), 298-308 (2015).
- Alvarez, S.W., Sviderskiy, V.O., Terzi, E.M., et al. Nature 551(7682), 639-643 (2017).
- Dixon, S.J., Winter, G.E., Musavi, L.S., et al. ACS Chem. Biol. 10(7), 1604-1609 (2015).
- Kagan, V.E., Mao, G., Qu, F., et al. Nat. Chem. Biol. 13(1), 81-90 (2017).
- Doll, S., Proneth, B., Tyurina, Y.Y., et al. Nat. Chem. Biol. 13(1), 91-98 (2017).
- Friedmann Angeli, J.P., Schneider, M., Proneth, B., et al. Nat. Cell Biol. 16(12), 1180-1191 (2014).
- Linkermann, A., Skouta, R., Himmerkus, N., et al. Proc. Natl. Acad. Sci. U.S.A. 111(47), 16836-16841 (2014).
- Zilka, O., Shah, R., Li, B., et al. ACS Cent. Sci. 3(3), 232-243 (2017).
- Shah, R., Margison, K., and Pratt, D.A. ACS Chem. Biol. 12(10), 2538-2545 (2017).
- Poon, J.-F. and Pratt, D.A. Acc. Chem. Res. 51(9), 1996-2005 (2018).
- Hinman, A., Holst, C.R., Latham, J.C., et al. PLoS One 13(8), e0201369 (2018).


 Assay Kit")
-Sulfoximine")



")


")
-HpETE-sn-glycero-3-PE")
-HETE-sn-glycero-3-PE")
-RSL3")

